The lab I work in, Tobin Sosnick's Lab at the University of Chicago studies protein folding and protein structure prediction. Sorry that I am going to skip over most of the stuff but the general feature I am going to focus on is predicting secondary structure.
Proteins form different secondary structure based on sterics, i.e. space occupation and also neighbor effects, how their neighbor's interactions effect them. Our lab use a statistically potential for this derived from a library of trimers culled from the PDB. It works pretty well and they have published papers predicting structures using it. However, I felt that maybe this library could be improved because obviously, the known structures don't contain all the possible angles available for a given sequence of amino acids or the library probably doesn't even contain all possible combinations of sequence. I thought a good way to remedy this would be to use molecular dynamics simulations. In this vain I attempted to see if I could predict the secondary structure of a 3 residues combination in a protein. I thought it would be easy. I could just simulate every 3 residue combination, I mean it is only 20^3 = 8000(20 amino acids, 3 residues).
I started with alanine tripeptide. I used my home computers and GROMACS because it is easy to script, runs fast and works great! Most of the peptides were simulated for 10-20 nanoseconds using the OPLS-AA forcefield with tip4p water model.

So if you don't know anything about ramachandran maps, click here. Well I expected that this Alanine tripeptide would look alpha helical. So many studies have been done on alanine and it has strong helical propensities. The angles of phi,psi space that correspond to the helical region are around -75ish(Phi), -50(Psi). As you can see the simulation did not reproduce any of this hahaha. Damnit.
So I figured that maybe it was that there was a length issue. That the forcefield in the simulation could not recapitulate the physical properties without more residues to interact. So I switched to glutamate (~ same helical propensity as alanine, i.e. alot) and ran four, five and 6 residue simulations.

4 GLU
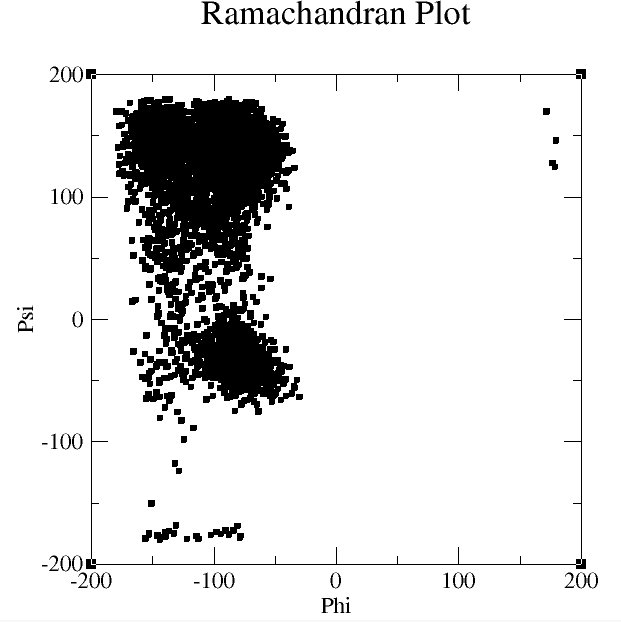
5 GLU

6 GLU
NICE! What we can see is that as we increase the number of residues the simulations becomes more and more like what would be predicted. Unfortunately I don't think there are many? Any? (Wel lto my surprise I just tried Blast and there are alot of proteins with 6 glutamates... Damn... haha).
Anyways, I wanted to try and predict a sequence that a secondary structure propensity was not well know or that the Sosnick lab could not predict well but one in which there was a structure in the PDB so I know if my prediction was correct or not. I random searched the PDB for EIYYINH because I wanted a sequence of at least 6 residues. Looking at the combination of 3 residue predictions of EIYYINH from the Sosnick server we see that it would mostly classify it as alpha helical with slight beta sheet preferences.

EIYYINH
My simulations however show a very strong beta sheet preference with other stuff throw in probably for the residues near the end of the chain. This what is in the structure!! Beta sheet structure generally has Phi, Psi angles in the -125ish(Phi), 150ish(Psi) region. http://www.rcsb.org/pdb/explore/remediatedSequence.do?structureId=2L4J
YAY!
Really cool.
Caveats: this does not man I can predict structure just the secondary structure preference of 6 residue regions of proteins. The hard part is actually folding it into a 3-dimensional structure. I was thinking of a sliding window protocol to simulate a protein and then see if it could pieced together with maybe throwing in some energy functions to combine it and fold it. That seems like alot of work and I don't feel like doing it. I am happy with my result and my hypothesis that maybe their trimer library could use more sampling.
No, I have not told them. I don't think they really care, hah. It was just for fun anyway and to test my idea.